Principales caractéristiques des anomalies génétiques du métabolisme du fer
Les anomalies du métabolisme du fer d'origine génétique surviennent chez des personnes porteuses de mutations sur certains gènes impliqués dans le métabolisme du fer.
| Gène | Transmission | Age | Expression | |
| HFE | HFE | Récessive | Adulte | Foie, articulations |
| Ferroportine | SLC40A1 | Dominante | Adulte | Peu de signes sauf si cofacteurs |
| L ferritine | L-FRT | Dominante | Adulte ± jeune | Cataracte précoce (inconstante) |
| Céruloplasmine | CP | Récessive | Adulte ± jeune | Cp effondrée. Anémie, s. neuro & diabète |
| Récepteur transferrine 2 | RTf2 | Récessive | Adulte | Foie, articulations (idem HFE) |
| Hémojuvéline | HJV | Récessive | Adulte jeune | Foie, diabète, cœur, gonades |
| Hepcidine | HAMP | Récessive | Adulte jeune | Foie, diabète, cœur, gonades |
SURCHARGES EN FER
Le fer, mal régulé, s’accumule dans différents organes, en particulier le foie, le pancréas, les articulations, les os ou le cœur voire le cerveau. Le fer est certes un élément indispensable à la vie mais en quantité trop importante il devient toxique. Il peut alors être à l’origine d’une cirrhose, d’un diabète, de rhumatismes, d’une ostéoporose, d’une insuffisance ante-hypophysaire, de troubles du rythme ou d’une insuffisance cardiaque voire de troubles neurologiques.
Surcharges liées à une sortie excessive de fer des cellules intestinales et macrophagiques.
Elles sont la conséquence d’une carence en hepcidine, hormone de régulation du fer, ou d’une insensibilité de la ferroportine, transporteur intracellulaire du fer, à l’hepcidine. Elles sont à l’origine d’une surcharge plus ou moins importante caractérisée par une augmentation du coefficient de saturation de la transferrine.
- L’hémochromatose de type 1 - dite hémochromatose HFE - est la plus fréquente et, à ce titre, ne répond pas stricto sensu à la définition d’une maladie rare. Toutefois, dans la mesure où sa pénétrance est faible, elle est intégrée aux surcharges considérées dans le cadre du CRFer. Liée à une anomalie du gène HFE, essentiellement la mutation C282Y, elle est de transmission récessive et s’exprime après 35 ans par une accumulation progressive du fer, essentiellement dans le foie.
- L’hémochromatose de type 2 dite juvénile est due à des mutations du gène de l’hémojuvéline(hémochromatose de type 2A) ou du gène de l’hepcidine (hémochromatose de type 2B). De transmission récessive, elle touche le sujet jeune en donnant une maladie en règle précoce et sévère.
- L’hémochromatose de type 3est due à des mutations du gène du récepteur 2 de la transferrine. De transmission récessive, elle est, dans sa forme habituelle, cliniquement indiscernable d’une hémochromatose de type 1. Cependant elle peut survenir chez le jeune adulte.
- L’hémochromatose de type 4B est due à certaines mutations du gène de la ferroportine qui rendent la protéine insensible à l’action de l’hepcidine. De transmission dominante, elle se présente comme une hémochromatose de type 1.

Hémochromatoses
’atteinte d’un des gènes impliqués dans la synthèse de l’hepcidine (HFE, HJV = hémojuvéline, RTf2 = récepteur de la transferrine 2, HAMP = hepcidine) aboutit à une carence plus ou moins marquée en hepcidine responsable d’une entrée excessive de fer dans la circulation. La capacité de la transferrine s’en trouve débordée, ce qui conduit à l’apparition de fer non lié à la transferrine. Ce fer libre pénètre très facilement dans les organes dont le foie, le cœur et le pancréas. Elle est à l’origine de la surcharge. PUBLISHCertaines mutations du gène de la ferroportine responsables d’une insensibilité de la ferroportine à l’hepcidine aboutissent au même tableau.
Surcharges liées à une rétention du fer par les cellules.
Elles se caractérisent par un coefficient de la transferrine bas, normal ou peu augmenté.
- L’hémochromatose de type 4Aest due à certaines mutations du gène de la ferroportine qui empêchent la protéine de migrer dans la cellule et donc d’exporter le fer vers le secteur extracellulaire. De transmission dominante, elle est à l’origine d’une surcharge touchant un type de cellules particulier, les macrophages.
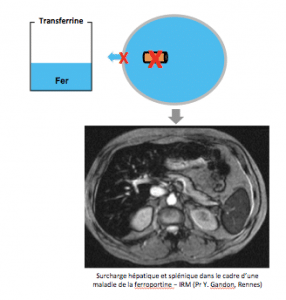
Maladie de la ferroportine Type 4 A
La ferroportine est un transporteur transmembranaire du fer qui permet la sortie de fer de la cellulaire. Certaines mutations du gène de la ferroportine aboutissent à la synthèse d’une protéine incapable de transporter le fer.
Il s’ensuit une accumulation de fer à l’intérieur de la cellule. Les macrophages qui sont particulièrement riches en ferroportine sont les premiers concernés par ce type de surcharge.
- L’acéruloplasminémie est une autre pathologie de surcharge en fer d’origine génétique, due à des mutations du gène de la céruloplasmine. De transmission autosomique récessive, elle se traduit par une absence de céruloplasmine dans le plasma et conduit à une accumulation de fer dans le foie et le cerveau.
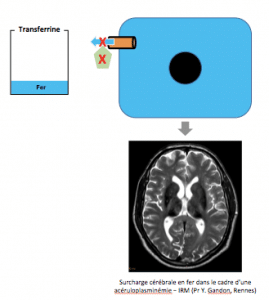
Acéruloplasminémie
La céruloplasmine est une protéine nécessaire à la sortie du fer de la cellule et à sa prise en charge par la transferrine. Certaines mutations du gène de la céruloplasmine sont responsables d’une absence de synthèse de céruloplasmine, laquelle n’est plus dosable dans le sang.
Le fer reste bloqué dans la cellule et s’y accumule. La particularité de cette surcharge est qu’elle concerne l’ensemble des cellules de l’organisme dont les cellules cérébrales.
- Des mutations du gène DMT1conduisent à une accumulation de fer au niveau du foie associée à une anémie microcytaire.
HYPERFERRITINÉMIES
Les hyperferritinémies génétiquespar mutation du gène de la L ferritine sont le fait de mutations du gène de la L ferritine. Elles sont transmises selon un mode autosomique dominant et ne s’accompagnent d’aucune surcharge en fer. Certaines s’expriment par une cataracte.
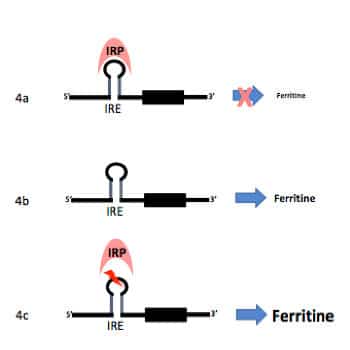
Hyperferritinémie génétique
L’ADN (= acide désoxyribonucléique qui constitue l’information génétique) est traduit en ARN (acide ribonucléique dont la lecture aboutit à la synthèse du produit du gène). Le système IRE-IRP (Iron Responsive Element sur l’ARN et Iron Responsive Protein dans la cellule) régule la dégradation de l’ARN en fonction de la position de l’IRE sur l’ARN.
Dans le cas de la ferritine, la liaison de l’IRP avec l’IRE empêche la lecture de l’ARN et donc la synthèse de ferritine (4a). Lorsqu’il n’est pas au contact de l’IRE, la lecture se fait sans obstacle et la ferritine est synthétisée (4b).
Lors des hyperferritinémies génétiques, une mutation de l’ARN de la ferritine vient bloquer ce système de régulation et il n’y a plus de frein à la synthèse de ferritine (4c).
POUR EN SAVOIR PLUS
Revues générales sur les surcharges en fer
- https://www.orpha.net/data/patho/Pub/fr/hemochromatose-FRfrPub92.pdf
- Encyclopédie Médico Chirurgicale 7-007-B-22
- Concours Médical
Focus sur différentes formes de surcharges en fer
- GeneReviews® - https://www.ncbi.nlm.nih.gov/books/NBK1116
- HFE-associated hereditary hemochromatosis, Seckington R & Powell LW
- TRfR2-related hereditary hemochromatosis, Camashella C & Roetto A
- Aceruloplasminemia, Miyajima H
- Orpha Net - https://www.orpha.net
- Hereditary haemochromatosis, Lancet
Focus sur les hyperferritinémies génétiques
Fiche de renseignements

Articles récents
-

Nouveau site internet du CRFER
Nouveau site internet pour le centre de Référen...